Прикладные пакеты
Здесь находятся рекомендации по установке, запуску и оптимизации для вычислений на кластере различных программных пакетов
Компиляция и запуск C2PK
C2PK — Программный пакет для квантовой химии и физики твердого тела. Репозиторий на github.
Последовательность сборки пакета в домашнем каталоге
#Выбор mpi - vapich4.1/gcc 8.5.0
module load mvapich4/gcc64
# Вспомогательная библиотека DBCSR от разработчиков C2PK
wget 'https://github.com/cp2k/dbcsr/releases/download/v2.9.1/dbcsr-2.9.1.tar.gz'
tar xf dbcsr-2.9.1.tar.gz
mkdir dbcsr-2.9.1/build
cd dbcsr-2.9.1/build
# каталог установки ~/dbcsr
cmake .. -DUSE_MPI=ON -DCMAKE_C_COMPILER=mpicc -DCMAKE_CXX_COMPILER=mpic++ -DCMAKE_Fortran_COMPILER=mpif90 -DCMAKE_INSTALL_PREFIX=${HOME}/dbcsr
make -j 8
make install
# Возвращаемся в исходный каталог
cd ../..
# Сборка CP2K
wget https://github.com/cp2k/cp2k/releases/download/v2026.1/cp2k-2026.1.tar.bz2
tar xf cp2k-2026.1.tar.bz2
mkdir cp2k-2026.1/build
cd cp2k-2026.1/build
# каталог установки ~/cp2k
cmake .. -DCMAKE_C_COMPILER=mpicc -DCMAKE_CXX_COMPILER=mpic++ -DCMAKE_Fortran_COMPILER=mpif90 -DCP2K_USE_MPI=ON -DCMAKE_INSTALL_PREFIX=${HOME}/cp2k
make -j 8
make install
cd ../..
Установка примеров (при желании):
git clone https://github.com/cp2k/cp2k-examples.git
Пример запуска из подкаталога _scratch/work домашнего каталога:
module load mvapich4/gcc64
export LD_LIBRARY_PATH="${HOME}/cp2k/lib64/:$LD_LIBRARY_PATH"
export PATH="${HOME}/cp2k/bin/:$PATH"
mkdir ~/_scratch/work
cd ~/_scratch/work
cp ~/cp2k-examples/gw/1_H2O_GW100/H2O_GW100_def2-QZVP.inp test.inp
export OMP_NUM_THREADS=8
export OMP_PLACES=cores
export OMP_PROC_BIND=true
srun --ntasks-per-node=2 --cpus-per-task=8 -N 1 cp2k.psmp -i test.inp > test.out
Оптимизация запуска LAMMPS
Сгенерировано ИИ
Выбор оптимального числа узлов кластера для LAMMPS — это процесс поиска баланса между скоростью счета и эффективностью использования ресурсов. Не существует единственного "правильного" числа, так как оно зависит от размера вашей системы, аппаратного обеспечения и физической модели. Вот пошаговая стратегия, которая поможет вам найти этот оптимум.
1. Проведите тест масштабирования
Это главный метод для определения лимитов масштабируемости. Суть в том, чтобы взять фиксированную задачу (вашу систему с определенным количеством атомов) и запускать её на нарастающем количестве ядер (например, N=1, 2, 4, 8, 16, 32, 64, 128...).
Что делать: Запустите вашу систему на 100-200 шагов для каждого количества ядер. Используйте команды balance или fix balance для распределения нагрузки .
На что смотреть:
- Время расчета (
Loop time): Сравните, во сколько раз ускорился расчет. Идеальное ускорение (100%) — это когда на 64 ядрах расчет идет в 64 раза быстрее. - Эффективность: Рассчитайте эффективность как
(T1 / TN) / N * 100%. Как правило, LAMMPS показывает эффективность >50%, пока на одно ядро приходится несколько сотен атомов .
Где предел: Вы увидите, что после определенного числа ядер время перестает значительно сокращаться. Это и есть точка насыщения, где накладные расходы на коммуникацию (обмен данными между узлами) начинают доминировать над вычислениями.
2. Используйте анализ профайлера (Timing Data)
После каждого прогона LAMMPS выводит детальную статистику. Это ваш главный инструмент диагностики. В конце файла log.lammps вы увидите таблицу:
| Section | %varavg | %total |
|---|---|---|
| Pair | ... | ... |
| Neigh | ... | ... |
| Comm | ... | ... |
| Other | ... | ... |
Следите за Comm (Communication): Если процент времени, потраченный на коммуникацию (Comm), при увеличении числа узлов становится слишком большим (например, >30-40%), дальнейшее добавление узлов неэффективно. Прирост производительности будет минимальным .
Следите за %varavg: Высокое значение (близкое к 100%) указывает на сильную несбалансированность нагрузки. Это значит, что одни ядра простаивают в ожидании других. В этом случае стоит сначала попробовать балансировку (fix balance) .
3. Учитывайте геометрию и разбиение (Domain Decomposition)
LAMMPS использует пространственное разбиение: симуляционный ящик режется на прямоугольные блоки, каждый из которых отдается одному MPI-процессу .
Проблема "блинов": Если вы используете "неудобное" число MPI-процессов (например, простое число, такое как 7 или 11), разбиение может получиться несбалансированным (например, длинные и тонкие "слайсы").
Что делать: Используйте команду processors в скрипте, чтобы вручную задать раскладку сетки (например, processors 4 4 4 для 64 ядер), чтобы блоки были максимально кубическими. Это минимизирует площадь поверхности обмена данными .
4. Учитывайте тип потенциала и системы
Разные задачи масштабируются по-разному :
Потенциалы малого радиуса (LJ, EAM): Обычно хорошо масштабируются. Ограничением часто становится сетка для дальнодействия (PPPM).
Дальнодействие (Kspace, PPPM): Если в системе есть кулоновские взаимодействия (вода, ионы), расчеты PPPM включают быстрые преобразования Фурье (FFT). FFT требует глобальной коммуникации, что сильно ухудшает масштабируемость за пределы одного узла. В таких системах "золотой серединой" часто является 1-4 узла, даже если у вас много ядер.
Связанные списки (Neigh): Если в профайлере Neigh занимает >50% времени, попробуйте увеличить параметр every в команде neigh_modify (например, с 1 на 10), чтобы реже перестраивать списки соседей .
5. Настройка гибридного параллелизма (MPI + OpenMP)
Для многоядерных узлов часто эффективнее использовать гибридный режим вместо "чистого" MPI.
Правило: Запускайте 1 MPI-процесс на сокет (физический чип) или на группу ядер, используя OpenMP для распараллеливания внутри узла.
Зачем: Это уменьшает количество MPI-сообщений, которыми обмениваются узлы, и снижает нагрузку на коммуникационную сеть .
Резюме: Пошаговый алгоритм действий
- Оцените размер: Для хорошей производительности на одно ядро должно приходиться от 500 до 10 000 атомов.
- Балансировка: Если система неоднородна (например, жидкость с пузырьками или мембрана), обязательно используйте
fix balance 10 1.1 rcb. - Тест: Начните с 1 узла, увеличивая количество ядер (но не выходя за пределы 500–1000 атомов/ядро).
- Проверка узлов: Когда эффективность на одном узле упадет ниже 70-80% (смотрите на рост
Comm), начните добавлять второй, третий и четвертый узлы. - Стоп-критерий: Как только время расчета перестает уменьшаться пропорционально количеству добавленных ядер (например, добавили 10 ядер, а расчет стал быстрее только на 5%), вы нашли оптимум.
Важное примечание
Официальная документация LAMMPS подчеркивает: "Нет замены определению узких мест производительности и опробованию различных вариантов" . Всегда проверяйте гипотезы короткими прогонами (100-200 шагов) на вашей конкретной системе и вашем конкретном кластере.
Параллельный Matlab
Система Matlab (Matrix Laboratory) - разработка компании The MathWorks, предназначенная для выполнения математических расчетов при решении научных и инженерных задач.
Достоинства Matlab – это, прежде всего, простота матричных операций и наличие многочисленных пакетов программ (Toolbox-ов), среди которых
Parallel Computing Toolbox, расширяющий Matlab на уровне языка операциями параллельного программирования.
Parallel Computing Toolbox достаточно для написания и запуска параллельной Matlab программы на локальной машине (Product Documentation).
Вычисления с Matlab на кластере требуют уже 2 продукта:
Parallel Computing Toolbox и
Matlab Distributed Computing Server
В ИММ имеются все 3 основных продукта для параллельных вычислений с Matlab на кластере "Уран" (версия Matlab R2011b и старше):
1) Matlab: 10 лицензий,
2) Parallel Computing Toolbox: 10 лицензий
(прежнее название Distributed Computing Toolbox),
3) Matlab Distributed Computing Server: 1000 лицензий
(прежнее название Matlab Distributed Computing Engine);
а также большое количество специализированных Toolbox-ов: по 10 лицензий на SIMULINK, Signal_Blocks, Image_Acquisition_Toolbox, Image_Toolbox, MAP_Toolbox, Neural_Network_Toolbox, Optimization_Toolbox, PDE_Toolbox, Signal_Toolbox, Statistics_Toolbox, Wavelet_Toolbox и 2 лицензии на Filter_Design_Toolbox.
Список всех установленных на кластере продуктов Matlab и количество доступных лицензий на них можно уточнить командой
/opt/matlab-R2010a/etc/lmstat -aНазвание текущей рабочей версии Matlab можно узнать, набрав, например, в командной строке
echo 'exit' | matlab -nodisplayили выполнив в окне Matlab команду ver.
Замечания.
1. При работе в Matlab следует ориентироваться на документацию используемой версии.
2. Для смены текущей версии следует воспользоваться командой module.
Можно запускать программы из командной строки или из системы Matlab.
Для запуска программ на кластере из командной строки пользователю необходимо установить на своем компьютере программу PuTTY.
Запуск параллельных Matlab-программ из командной строки осуществляется с помощью разновидностей команды mlrun.
Для работы в диалогом окне Matlab на компьютере пользователя предварительно должен быть установлен и запущен какой-нибудь X-сервер (MobaXterm, X2Go).
Из командной строки вызвать Matlab
matlab или
matlab & (с освобождением командной строки)
и дождаться появления оконного интерфейса Matlab.
Запуск параллельных программ на кластере из системы Matlab осуществляется с помощью разновидностей служебной функции imm_sch.
Замечание. Можно работать в системе Matlab, запустив её в интерактивном текстовом режиме командой
matlab -nodisplayВыход осуществляется по команде exit.
Параллельные вычисления на кластере инициируются
1) запуском параллельных програм;
2) запуском частично параллельных программ (c parfor или spmd);
3) запуском программ с использованием GPU.
Подробнее на нижеследующих страницах.
В разделе "Запуск задач на кластере" находится краткая инструкция "Запуск параллельного Matlab".
Использование русских букв в Linux версии Matlab
Для работы с русскими буквами в Matlab'е необходимо правильно настроить кодировку файла с программой, и, при необходимости, настроить ввод русских букв в клиентской программе.
Возможны два варианта настройки кодировки файла с программой:
- Если в основном вы работаете в Windows, то рекомендуется использование кодировки CP1251.
- Если вы создаёте, редактируете и запускаете программы в основном в Linux, то рекомендуется использование кодировки UTF-8 (кодировка на кластере по умолчанию).
Вариант 1
На кластере "Уран" при запуске Matlab'а можно включить Windows-кодировку CP1251. Для этого необходимо запускать Matlab в окне терминала следующей командой:
LANG=ru_RU.CP1251 matlabПосле этого можно нормально работать с файлами, подготовленными в Windows.
Внимание. Данный вариант может не сработать при запуске счётной задачи на узлах кластера.
Как минимум, в начало счетной программы надо вставить команду:
feature('DefaultCharacterSet', 'cp-1251');
После этого строки, выводимые функцией fprintf(), будут сохраняться в правильной кодировке. Строки на русском языке, выводимые функцией disp(), к сожалению, будут испорчены в любом случае.
Если в программе для вывода результатов используется функция disp(), выполняются операции сравнения или сортировки строк с русскими буквами, то стоит попробовать второй вариант.
Вариант 2
После передачи файла из Windows на кластер можно перекодировать его в кодировку UTF-8. Следует помнить, что перекодированный файл будет некорректно отображаться в Windows, зато он без проблем будет обрабатываться на кластере.
2.a Перекодирование файла на кластере в командной строке. Файл перекодируется с помощью команды
iconv -c -f WINDOWS-1251 -t UTF-8 winfile.m > unixfile.m где winfile.m - имя исходного файла, а unixfile.m - перекодированного (подставьте вместо winfile.m и unixfile.m имена ваших файлов, главное, помните - они должны быть различными). Затем можно открыть новый файл в Matlab'е.
2.b Перекодирование файла на кластере в текстовом редакторе. Для перекодирования файла необходимо запустить текстовый редактор KWrite (Linux'овская кнопка "Пуск", затем ввести в строке поиска имя редактора). При открытии файла с программой указать кодировку cp1251, убедиться, что русские буквы читаются правильно, затем выбрать пункт меню "Файл ->Сохранить как" и указать при сохранении кодировку UTF-8.
Если есть необходимость перекодировать файлы, полученные с кластера, то это также можно сделать с помощью KWrite. При открытии файла надо выбрать кодировку UTF-8, а потом сохранить файл в кодировке cp1251.
В командной строке перекодирование из Linux в Windows выглядит так:
iconv -c -f UTF-8 -t WINDOWS-1251 unixfile.m > winfile.m
Примечание для администраторов
В дистрибутиве RHEL и его производных (CentOS, Scientific Linux) отсутствует файл локализации ru_RU.CP1251. Поэтому "Вариант 1" не сработает (Matlab не запустится с сообщением о невозможности установить указанный язык).
Для генерации файла с кодировкой администратор должен выполнить в Linux'е команду:
localedef -i ru_RU -f CP1251 ru_RU.CP1251
Историческое примечание
Старые версии Matlab'а (до 2010 года) умели работать с единственной кодировкой русских букв ISO-8859-5. Для того, чтобы ее настроить на серверной стороне, необходимо установить переменную окружения LANG в значение ru_RU.8859-5 .
1) Для корректной обработки русских букв на вычислительных узлах
в файл ".bashrc" из домашнего каталога пользователя (~/.bashrc) необходимо вставить следующую строку:
export LANG=ru_RU2) в настройках сессии программы PuTTY, в разделе Translation, установить кодировку ISO-8859-5:1999 (Latin/Cyrillic) и сохранить эту сессию для работы с системой Matlab в дальнейшем.
Запуск параллельной программы
Основные сведения
Параллельная программа - это программа, копии которой, запущенные на кластере одновременно, могут взаимодействовать друг с другом в процессе счета.
Программа пользователя должна быть оформлена как функция (не скрипт) и находиться в начале запускаемого файла, т.е. предшествовать возможным другим вспомогательным функциям. Имя файла должно совпадать с именем первой (основной) функции в файле. Одноименная с файлом функция, не являющаяся первой, никогда не будет выполнена, так как независимо от имени всегда выполняется первая функция файла. Файл должен иметь расширение "m" (Пример параллельной программы).
Для выполнения программы пользователя всегда вызывается программа MatLab.
При запуске программы пользователя на кластере в программе MatLab создаётся объект Job (работа) с описанием параллельной работы, которое включает определение объекта Task (задача), непосредственно связанного с заданной программой. Можно сказать, что копия работающей программы представлена в системе MatLab объектом Task.
Каждый объект Job получает идентификатор (ID) в системе MatLab, равный порядковому номеру. Нумерация начинается с 1.
Соответствующее имя работы вида Job1, выдаваемое при запуске, хранится в переменной окружения MDCE_JOB_LOCATION и может быть использовано в программе, а сам объект доступен пользователю во время сеанса MatLab.
Аналогично пользователь имеет доступ и к объектам Task (задача), которые также нумеруются с 1. Идентификатор или номер задачи (1,2,...) - это номер соответствующего параллельного процесса (lab) и его можно узнать с помощью функции labindex, а общее число запущенных копий с помощью функции numlabs.
Имена работы (Job1), задач (Task1, Task2,…) используются в процессе вычислений для формирования имен файлов и каталогов, связанных с заданной программой. Так, каталог вида Job1 содержит наборы файлов с информацией по задачам. Имена этих файлов начинаются соответственно с Task1, Task2, … Например, файлы вывода имеют вид Task1.out.mat, Task2.out.mat, … и содержат, в частности, выходные параметры функции пользователя (массив ячеек argsout).
Программа пользователя, оформленная в виде объекта Job, поступает в распоряжение системы запуска, которая ставит её в очередь на счет с присвоением своего уникального идентификатора.
Система запуска создает каталог вида my_function.1, например для файла my_function.m. В этом каталоге пользователю может быть интересен, в частности, файл errors (см. Возможные ошибки). Заметим, что пользователь должен сам удалять ненужные каталоги вида имя_функции.номер (номера растут, начиная с 1).
Если ресурсов кластера достаточно, то на каждом участвующем в вычислении процессоре (ядре для многоядерных процессоров) начинает выполняться копия программы-функции пользователя при условии наличия достаточного числа лицензий (в настоящее время система запуска не контролирует число лицензий, доступность лицензий определяется в начале счета).
Пользователь может контролировать прохождение своей программы через систему запуска как в окне системы Matlab, например, с помощью Job Monitor (см. п. Доступ к объекту Job ), так и из командной строки с помощью команд системы запуска (запросить информацию об очереди, удалить стоящую в очереди или уже выполняющуюся программу).
Действия пользователя
Войти на кластер (с помощью PuTTY или MobaXterm) и запустить программу-функцию из командной строки или в окне системы Matlab, указав необходимое для счета число параллельных процессов и максимальное время выполнения в минутах.
В ответ пользователь должен получить сообщение вида:
Job output will be written to: /home/u1303/Job1.mpiexec.out где Job1 - имя сформированной работы, 1 - идентификатор работы
(/home/u1303 - домашний (личный) каталог пользователя).
Замечание. Для локализации результатов вычислений рекомендуется осуществлять запуск программы (даже в случае запуска встроенных функций Matlab ) из рабочего каталога, специально созданного для данной программы в домашнем каталоге.
Запуск параллельной программы из командной строки
Команда запуска mlrun имеет вид
mlrun -np <number_of_procs> -maxtime <mins> <func> ['<args>']где<number_of_procs> - число параллельных процессов (копий программы)<mins> - максимальное время счета в минутах<func> - имя файла с одноименной функцией (например, my_function)<args> - аргументы функции (не обязательный параметр) берутся в одиночные кавычки и представляются в виде
k,{arg1,...,argn}
где k - число выходных аргументов функции, а в фигурных скобках список ее входных аргументов. При отсутствии аргументов у функции, что соответствует "0,{}", их можно опустить. Например, для файла my_function.m с одноименной функцией без параметров запуск имеет вид:
mlrun -np 8 -maxtime 20 my_functionгде 8 - число процессов, 20 - максимальное время счета в минутах.
Запуск параллельной программы в окне Matlab
В командном окне Matlab (Command Window) вызвать служебную функцию imm_sch, которой в качестве параметров передать число процессов, время выполнения и предназначенную для параллельных вычислений функцию с аргументами или без, сохраняя (рекомендуется) или не сохраняя в переменной (например, job) ссылку на созданный объект Job (имя функции набирается с символом "@" или в одиночных кавычках):
job = imm_sch(np,maxtime,@my_function,k {arg1,...,argn});или
job = imm_sch(np,maxtime,'my_function',k,{arg1,...,argn});Так, для примера выше запуск в окне Matlab будет иметь вид:
job = imm_sch(8,20,@my_function);Доступ к объекту Job, состояние работы
Все работы хранятся на кластере. При необходимости доступа к работе, на которую в текущий момент отсутствует ссылка, можно (1) в окне Job Monitor правой кнопкой мыши выделить нужную работу и выбрать соответствующую опцию в контекстном меню или (2) по идентификатору (ID) определить ссылку на работу (обозначенную ниже job), используя, например, команды:
c = parallel.cluster.Generic
job = c.findJob('ID',1)Состояние работы (State) можно:
(1) увидеть в окне Job Monitor или
(2) выдать в окне Command Window, набрав
job.State
Основные значения состояния работы следующие:
pending (ждет постановки в очередь)
queued (стоит в очереди)
running (выполняется)
finished (закончилась)
Окончания счета (состояние finished) можно ждать с помощью функции wait, wait(job) или job.wait()
Примечание.
Вышеприведенные команды предназначены для версий MatLab с профилем кластера, т.е. начиная с R2012a. В ранних версиях (с конфигурацией кластера) следует набирать:
s = findResource('scheduler', 'type', 'generic')
job = findJob(s,'ID',1)При этом в поле DataLocation структуры s должен быть текущий каталог (тот, в котором ищем работу). Если каталог другой, то можно выполнить
clear allи повторить предыдущие команды.
В окне Matlab (с R2012a) для работы job и любой ее задачи (Task) с номером n=1,2,... можно выдать (далее для удобства n=1)
1) протокол сеанса:
job.Tasks(1).Diary2) информацию об ошибках (поле ErrorMessage):
job.Tasks(1) или job.Tasks(1).ErrorMessage
3) результаты (значения выходных параметров) работы job в целом (по всем Task-ам):
out = job.fetchOutputsТогда для 1-ой задачи (Task) значение единственного выходного параметра:
res = out{1}
В случае нескольких выходных параметров соответственно имеем для 1-го, 2-го, ... :
res1 = out{1,1}
res2 = out{1,2}
Другой способ выдачи результатов по задачам
out1 = job.Tasks(1).OutputArgumentsЗамечания.
1. Эту же информацию можно выдать, запустив Matlab в интерактивном текстовом режиме (matlab -nodisplay) и набирая затем упомянутые команды.
2. Если ссылка на работу (job) отсутствует, то ее можно найти по идентификатору.
3. Для ранних версий (до R2012a) следует использовать следующие команды, чтобы выдать
1) протокол сеанса:
job.Tasks(1).CommandWindowOutput 3) результаты (значения выходных параметров) работы job в целом (по всем Task-ам):
out = job.getAllOutputArguments1. Начать работу на кластере рекомендуется с запуска своей последовательной программы в тестовом однопроцессном варианте, например,
mlrun -np 1 -maxtime 20 my_functionгде my_function – функция без параметров, максимальное время счета 20 минут.
2. После преобразования последовательной программы в параллельную её работоспособность можно проверить, выполняя шаги, приведенные в пункте Как убедиться в работоспособности программы при рассмотрении примеров с распределенными массивами.
Пример запуска программы
Для запуска программы на кластере используем функцию rand, вызываемую для генерации 2х3 матрицы случайных чисел (традиционный вызов функции: y = rand(2,3)).
Для получения 4 экземпляров матрицы задаем число процессов, равное 4. Максимальное время счета пусть будет равно 5 минутам.
Запускать функцию будем на кластере "Уран" (umt) в каталоге test, специально созданном заранее в домашнем каталоге пользователя /home/u9999, где u9999 – login пользователя (см. Схема работы на кластере и Базовые команды ОС UNIX).
Для запуска функции rand из командной строки используется команда mlrun,
а из системы Matlab – служебная функция imm_sch.
Для запуска из командной строки войти на umt через PuTTY и выполнить команду
mlrun -np 4 -maxtime 5 rand '1,{2,3}'Для запуска из окна Matlab войти на umt из MobаXterm, вызвать Matlab командой
matlab &и в открывшемся окне набрать
job = imm_sch(4,5,@rand,1,{2,3});где job – ссылка на сформированную работу.
Можно работать в системе Matlab, войдя на umt через PuTTY и запустив её в интерактивном текстовом режиме командой
matlab -nodisplayВ ответ на приглашение (>>) следует соответственно набрать
job = imm_sch(4,5,@rand,1,{2,3});Выход из Matlab осуществляется по команде exit.
После выполнения mlrun или imm_sch выдается строка вида
Job output will be written to: /home/u9999/test/Job1.mpiexec.outгде Job1 - имя сформированной работы, 1 - идентификатор (номер) работы.
Если ресурсов кластера достаточно, задача войдет в решение (см. Запуск задач на кластере). Иначе для ускорения запуска на кластере небольших (отладочных) задач можно вместо выделенного задаче раздела назначить debug командой вида:
scontrol update job 8043078 partition=debugгде 8043078 — уникальный идентификатор (JOBID) задачи.
Дожидаемся окончания задачи, т.е. job.State должно быть finished.
В окне Matlab контролировать состояние задачи удобно с помощью Job Monitor.
Результаты выдаем с помощью команд:
со всех процессов
out = job.fetchOutputsа для выдачи матрицы, полученной 1-ым процессом (т.е. Task1)
out{1}и т.д.
Для выдачи результатов ранее посчитанной работы, на которую в текущий момент нет ссылки, следует обеспечить к ней доступ, например используя Job Monitor.
Запуск частично параллельной программы (c parfor или spmd)
- Введение
- 1. Запуск частично параллельной программы с неявным заданием пула
- 2. Запуск частично параллельной программы с явным заданием пула
на основе профиля кластера
Введение
Частично параллельной будем называть программу, при выполнении которой наряду с последовательными возникают параллельные вычисления, инициируемые параллельными конструкциями языка Matlab parfor и spmd.
Использование параллельного цикла parfor или параллельного блока spmd предполагает предварительное открытие Matlab пула, т.е. выделение необходимого числа процессов (Matlab workers или labs), на локальной машине или на кластере.
Запуск частично параллельных программ с открытием Matlab пула на кластере можно выполнять
(1) по аналогии с запуском параллельных программ из командной строки или в окне системы Matlab (запуск с неявным заданием пула) или
(2) на основе профиля кластера (с версии R2012a, ранее параллельной конфигурации) при работе в окне системы Matlab (запуск с явным заданием пула).
Пользователь может контролировать прохождение своей программы через систему запуска как в окне системы Matlab (с версии R2011b) с помощью Job Monitor (см. пункт меню Parallel), так и из командной строки с помощью команд системы запуска.
1. Запуск частично параллельной программы с неявным заданием пула
При запуске из командной строки вместо команды mlrun (для параллельных программ) следует использовать команду mlprun, например,
mlprun -np 12 -maxtime 20 my_function '1, {x1, x2}'а при запуске в окне вместо функции imm_sch использовать функцию imm_sch_pool, например,
job = imm_sch_pool(12,20,@my_function,1,{x1,x2});для функции, определенной как
function y = my_function(x1,x2)и запущенной на 12 процессах с максимальным временем счета 20 минут.
При этом один процесс будет выполнять программу-функцию, а оставшиеся будут использованы в качестве пула.
В результате запуска программа ставится в очередь на счет и, если ресурсов кластера достаточно, входит в решение.
Вывод результатов осуществляется так же, как и в случае параллельных программ.
2. Запуск частично параллельной программы с явным заданием пула
на основе профиля кластера
Явное задание пула возможно при работе в окне Matlab с помощью команды matlabpool (см. help matlabpool). Число выделенных процессов и время, в течение которого они будут доступны пользователю, зависят от заданного профиля кластера (см. пункт меню Help/Parallel Computing Toolbox/Cluster Profiles).
Команда
matlabpoolбез параметров открывает пул, используя профиль по умолчанию с указанным в нем размером пула. В ИММ УрО РАН по умолчанию Matlab пул открывается на узлах кластера (тип кластера Generic), поскольку управляющий компьютер, выступающий в роли локальной машины (Matlab client), не должен использоваться для длительных вычислений.
Пользователь может выбрать профиль по умолчанию из уже существующих профилей или создать новый, используя пункт меню Parallel окна Matlab.
Команда matlabpool с указанием размера пула, например
matlabpool open 28открывает пул, переопределяя размер, заданный по умолчанию. При этом следует иметь в виду, что существует ограничение на максимальное число доступных пользователю процессов на кластере.
В результате открытия пула сформированная для кластера работа с именем вида JobN (где N=1,2,...) поступает в распоряжение системы запуска и ставится в очередь на счет. Если свободных процессов достаточно, то пул будет открыт на время, заданное в профиле по умолчанию, с выдачей сообщения вида:
Starting matlabpool using the 'imm_20mins' profile ... Job output will be written to: /home/u1303/my_directory/Job1.mpiexec.out
connected to 28 labs.Размер пула можно узнать, набрав
matlabpool sizeПо завершении вычислений, связанных с пулом, его следует закрыть
matlabpool closeВнимание. Пул закрывается по истечении времени с диагностикой вида:
The client lost connection to lab 12.
This might be due to network problems, or the interactive matlabpool job might have errored.
Запуск частично параллельной программы на Matlab клиенте
(не разрешается для длительных вычислений)
Итак, схема использования параллельных конструкций parfor и spmd в окне Matlab такова:
matlabpool
...
% вычисления с использованием parfor или spmd,
% выполняемые построчно или
% собранные в программу (частично параллельную)
% и запущенные из файла
...
matlabpool closeПри этом все вычисления, кроме параллельных, выполняет Matlab client (см., например, Introduction to Parallel Solutions/Interactively Run a Loop in Parallel.)
Запуск частично параллельной программы с помощью команды batch
В системе Matlab существует команда (функция) batch, которая позволяет запускать программы в пакетном режиме, разгружая Matlab client (см., например, Introduction to Parallel Solutions/Run a Batch Job). Эта команда выполняется асинхронно, т.е. интерактивная работа пользователя не блокируется.
Где будет выполняться программа и максимально сколько времени, определяется планировщиком (scheduler), заданным в профиле кластера по умолчанию: в ИММ на кластере, тип планировщика generic. Для выполнения программы создается объект Job (работа). Команда batch вида
job = batch('my_mfile')(job - ссылка на объект работа)
запускает программу (скрипт или функцию) my_mfile в однопроцессном варианте. В ответ на команду batch выдается сообщение вида
Job output will be written to: /home/u1303/my_directory/Job1.mpiexec.outсодержащее идентификатор работы (ID), равный здесь 1. Сформированная для кластера работа ставится в очередь и при наличии свободных процессов входит в решение с именем очереди вида my_mfile.1.
Для запуска на кластере частично параллельной программы можно использовать команду batch с открытием пула. При этом выделяемое на кластере число процессов будет на 1 больше заданного размера пула.
Так, например, для выполнения команды
job = batch('my_mfile','matlabpool',11)потребуется 12 процессов: 1 для программы my_mfile, 11 для пула (см., например, Introduction to Parallel Solutions/Run a Batch Parallel Loop). Время счета здесь определяется временем, заданным в профиле по умолчанию (в аналогичной команде imm_sch_pool все аргументы задаются явно).
По завершении работы job можно выдать
1) протокол сеанса
job.diary2) результаты работы
out=job.fetchOutputs
celldisp(out)
(fetchOutputs вместо getAllOutputArguments в ранних версиях, использовавших конфигурацию кластера, а не профиль)
3) информацию об ошибках (поле ErrorMessage)
job.Tasks(1)или
job.Tasks(1).ErrorMessageЗапуск программ с использованием GPU
Программа с использованием GPU - это параллельная программа, каждая ветвь (копия, процесс), которой может использовать своё GPU.
Поэтому к программе с GPU применимо почти все, что относится к параллельной программе (в частности, это должна быть программа-функция). Особенности запуска программ с GPU отмечены ниже.
Программа с использованием GPU в результате запуска ставится в очередь на счет в системе SLURM. Пользователь может контролировать прохождение своей программы через эту систему с помощью соответствующих команд или в окне системы Matlab (с версии R2011b) с помощью Job Monitor (см. пункт меню Parallel).
При запуске программы-функции, как обычно, указывается необходимое для счета число параллельных процессов и максимальное время выполнения в минутах, но используются другие команды.
Запуск программы на счет осуществляется по аналогии с запуском параллельных программ:
из командной строки с помощью команды mlgrun, например,
mlgrun -np 8 -maxtime 20 my_gpufunction
или в окне системы Matlab с помощью служебной функции imm_sch_gpu, например,
job = imm_sch_gpu(8,20,@my_gpufunction);
В приведенных командах запускается функция без параметров с использованием 8 процессов, 20 минут - максимальное время счета.
При этом каждый из 8 процессов (копий функции) может использовать своё GPU.
При наличии параметров у функции они указываются по тем же правилам, что и в случае параллельной программы.
Возможные ошибки
1) Если не хватает лицензий для запуска на кластере функции my_function из одноименного файла my_function.m, то информация об ошибке находится в файле вида my_function.1/errors. При этом, как правило, файл my_function.1/output содержит MPIEXEC_CODE=123. Число свободных лицензий, кто и какие лицензии занимает, можно узнать, используя команду lmstat.
Внимание. В настоящее время система запуска не контролирует число лицензий, доступность лицензий определяется только в начале счета.
2) ErrorMessage содержит ошибки трансляции, например:
Undefined function or variable 'my_function'.Имя или расширение файла, имя функции или переменной отсутствует или указано неверно. Имя может отсутствовать по причине случайного запуска из другого каталога.
Invalid function name 'j-cod'.В именах файлов и функций не должно быть минуса ("-"), только подчерк ("_").
Invalid file identifier. Use fopen to generate a valid file identifier.Возможно, имя файла задано русскими буквами.
Внимание. В случае таких ошибок в файле my_function.1/errors обычно содержится строка
[0]application called MPI_Abort(MPI_COMM_WORLD, 42) - process 0Помните: имя файла должно совпадать с именем первой функции в файле, так как
(1) при запуске программы ищется файл с именем, указанным в команде запуска, и
(2) в нем выполняется, прежде всего, первая функция.
Если они не совпадают и при запуске указано имя функции (например, my_function), то файл не будет найден и будет выдана ошибка вида:
Undefined function or variable 'my_function'. Если они не совпадают и при запуске указано имя файла (например, my_code), то выполнится первая функция файла независимо от ее имени. При этом наличие одноименной с файлом функции, не являющейся первой, приведет к выдаче в протоколе сеанса предупреждения вида:
Warning: File: my_code.m Line: 25 Column: 14
Function with duplicate name "my_code" cannot be called.
3) Если выполнение программы прервано принудительно, например, по истечению времени, то выходная информация отсутствует (т.е. файлы вывода вида Task1.out.mat будут пусты). При этом состояние работы (поле State) будет running, в то время как состояния её задач (поля State для Tasks(n), где n=1,2,...) могут быть как running, так и finished, если часть копий программы успела финишировать до окончания заказанного времени.
В случае исчерпания времени соответствующая информация попадает в файл my_function.1/errors.
4) В случае аварийного завершения работы программы в домашнем каталоге пользователя (~) могут оставаться файлы вида mpd.hosts.123456 и mpd.mf…
Их следует периодически удалять вручную.
Завершение работы
Работу, в которой нет больше необходимости, следует уничтожить, используя Job Monitor (Delete в контекстном меню) или функцию delete, освобождая тем самым ресурсы кластера
job.deleteтакже удалить файл вида Job1.mpiexec.out (где Job1 - имя удаляемой работы),
а затем почистить Workspace
clear job
Если эти действия не выполняются пользователем регулярно, то при очередных запусках будут создаваться и накапливаться файлы новых работ Job2, Job3 и т.д.
Не все работы заканчиваются с признаком finished. Так, по истечении времени счета работа будет прервана в состоянии running.
Для уничтожения в текущем каталоге всех или только завершившихся (finished) работ можно воспользоваться написанной в ИММ УрО РАН функцией job_destroy.
Вызов job_destroy без параметра
из командной строки:
echo 'job_destroy, exit' | matlab -nodisplayв окне:
job_destroyуничтожает только завершившиеся работы.
Вызов job_destroy с параметром (тип и значение параметра не существенны)
из командной строки:
echo 'job_destroy(1), exit' | matlab -nodisplayили
echo "job_destroy('all'), exit" | matlab -nodisplayв окне:
job_destroy(1)или
job_destroy('all')уничтожает все работы в текущем каталоге.
Внимание! В состоянии running, разумеется, находятся выполняющиеся в текущий момент работы, поэтому выполняйте команду job_destroy(1) только тогда, когда Вы твердо уверены, что все работы закончились (нормально или аварийно).
После выполнения этой команды нумерация работ начинается с 1.
После уничтожения ненужных работ следует удалить на них ссылки в Workspace с помощью команды clear.
Важные замечания.
1. Нумерация работ (Job) в каталоге пользователя начнется с 1 в новом сеансе Matlab при отсутствии каталогов и файлов предыдущих работ (Job...).
2. Каталог вида my_function.1 не удаляется при использовании команды job_destroy. При новых запусках одной и той же программы образуются аналогичные каталоги с возрастающими номерами: my_function.2, my_function.3…
Пользователь должен сам удалять ненужные каталоги.
Пример параллельной программы
Пример взят с сайта MathWorks и иллюстрирует основные (базовые) принципы программирования параллельной программы-функции.
Копия работающей функции, т.е. задача (Task), для которой значение labindex равно 1, создает магический квадрат (magic square) с числом строк и столбцов равным числу выполняющихся копий (numlabs) и рассылает матрицу с помощью labBbroadcast остальным копиям. Каждая копия вычисляет сумму одного столбца матрицы. Все эти суммы столбцов объединяются с помощью функции gplus, чтобы вычислить общую сумму элементов изначального магического квадрата.
function total_sum = colsum
if labindex == 1
% Send magic square to other labs
A = labBroadcast(1,magic(numlabs))
else
% Receive broadcast on other labs
A = labBroadcast(1)
end
% Calculate sum of column identified by labindex for this lab
column_sum = sum(A(:,labindex))
% Calculate total sum by combining column sum from all labs
total_sum = gplus(column_sum)
endСуществуют альтернативные методы получения данных копиями функций, например, создание матрицы в каждой копии или чтение каждой копией своей части данных из файла на диске и т.д.
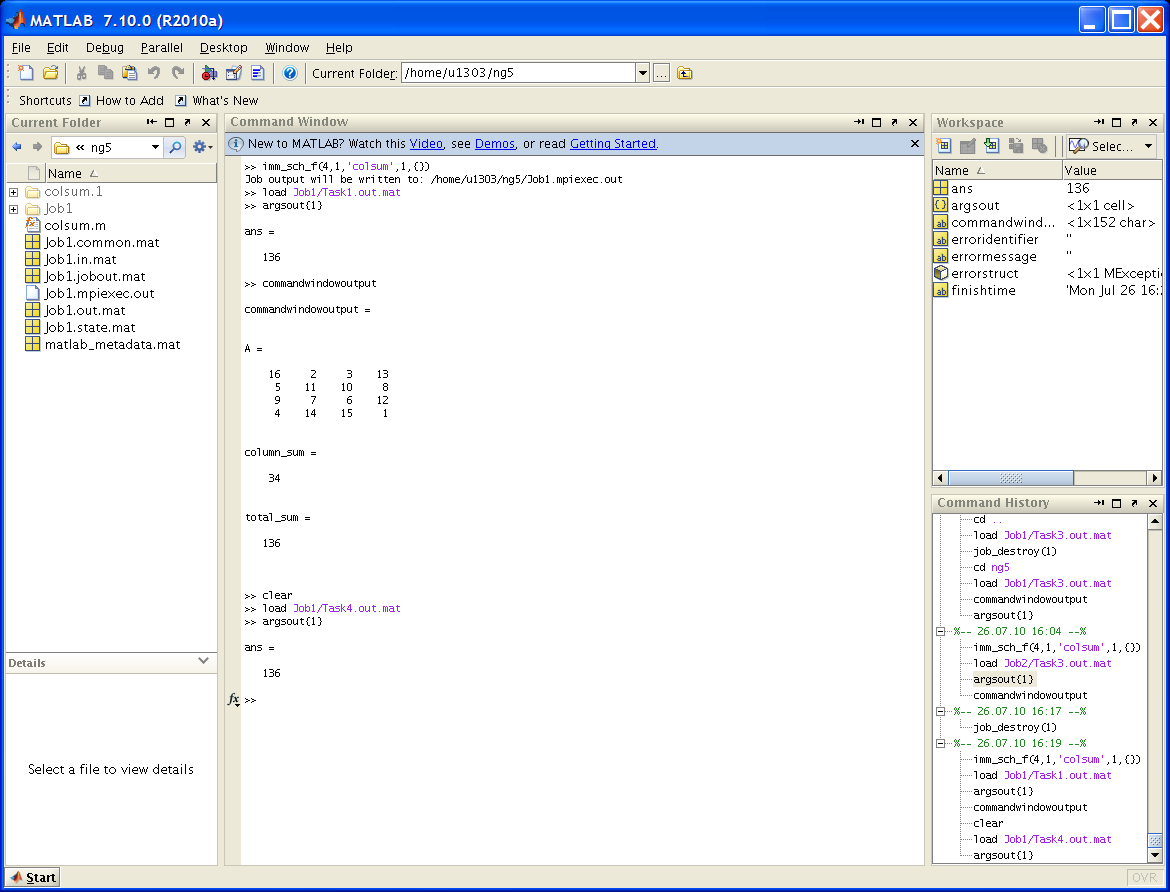
В прикрепленном файле ml_session.PNG (см. ниже) демонстрируется запуск функции colsum в окне системы Matlab с использованием 4-х вычислительных процессов и максимальным временем счета 1 минута.
Функция хранится как файл colsum.m в каталоге ng5 пользователя (полный путь: /home/u1303/ng5). После запуска imm_sch_f(4,1,'colsum',1,{});, а в более поздних версиях (с учетом переименования служебной функции в imm_sch)
job = imm_sch(4,1,@colsum,1);выдается сообщение вида:
Job output will be written to: /home/u1303/ng5/Job1.mpiexec.outв котором указано имя сформированной работы Job1.
Проверив состояние работы и убедившись, что она закончилась, можно выдать результаты счета.
| Прикрепленный файл | Размер |
|---|---|
| 73.66 КБ |
{kind=link}
Примеры с распределенными массивами
Введение
Распределение данных по процессам предназначено для ускорения счета и экономии памяти.
Параллельные вычисления с использованием распределенных массивов подробно описаны на сайте Matlab, в частности, в подразделе Working with Codistributed Arrays.
Распределенный массив состоит из сегментов (частей), каждый из которых размещен в рабочей области (workspace) соответствующего процесса (lab). При необходимости информацию о точном разбиении массива можно запросить с помощью функции getCodistributor (подробнее см. Obtaining information About the Array). Для просмотра реальных данных в локальном сегменте распределенного массива следует использовать функцию getLocalPart.
Возможен доступ к любому сегменту распределенного массива.
Доступ к локальному сегменту будет быстрее, чем к удаленному, поскольку последний требует посылки (функция labSend) и получения (функция labReceive) данных между процессами.
Содержимое распределенного массива можно собрать с помощью функции gather в один локальный массив, продублировав его на всех процессах или разместив только на одном процессе.
Использование распределенных массивов при параллельных вычислениях сокращает время счета благодаря тому, что каждый процесс обрабатывает свою локальную порцию исходного массива (сегмент распределенного массива).
Существует 3 способа создания распределенного массива (см. Creating a Codistributed Array):
1) деление исходного массива на части,
2) построение из локальных частей (меньших массивов),
3) использование встроенных функций Matlab (типа rand, zeros, ...).
1) Деление массива на части может быть реализовано с помощью функции codistributed (Пример 1). При этом в рабочей области каждого процесса расположены исходный массив в своем полном объеме и соответствующий сегмент распределенного массива. Таким образом, этот способ хорош при наличии достаточного места в памяти для хранения тиражируемого (replicated) исходного массива.
Размерности исходного и распределенного массивов совпадают.
Деление массива может быть произведено по любому из его измерений.
По умолчанию в случае двумерного массива проводится горизонтальное разбиение, т.е. по столбцам, что выглядит естественно с учетом принятого в системе Matlab размещения матриц в памяти по столбцам (как в Фортране).
2) При построении распределенного массива из локальных частей в качестве сегмента распределенного массива берется массив, хранящийся в рабочей области каждого процесса (Пример 2). Таким образом, распределенный массив рассматривается как объединение локальных массивов. Требования к памяти в этом случае сокращаются.
3) Для встроенных функций Matlab (типа rand, zeros, ...) можно создавать распределенный массив любого размера за один шаг (см. Using MATLAB Constructor Functions).
Вверх
Как можно использовать распределенные массивы
В приводимых ниже примерах решение задачи связано с вычислением значений некоторой функции, обозначенной my_func. При этом вычисление одного значения функции требует длительного времени.
Даются возможные схемы решения таких задач с использованием распределенных массивов. Показан переход от исходной последовательной программы к параллельной с распределенными вычислениями. Соответствующие изменения выделены.
Замечание.
Хотя дистрибутивные массивы совместимы (в отличие от цикла parfor и GPU) с глобальными переменными, необходимо тщательно следить за тем, чтобы изменения глобальных переменных в процессе обработки одной порции данных не влияли на результаты обработки других порций, тем самым обеспечивая независимость вычислений частей дистрибутивных массивов.
Там, где допустимо, проще использовать цикл parfor или вычисления на GPU.
Вверх
Пример 1. Деление массива на части
Пусть требуется вычислить значения некоторой функции 10 вещественных переменных. Аргументом функции является 10-мерный вектор.
Значение функции необходимо вычислить для n точек (значений аргумента), заданных 10хn матрицей. Вычисление матрицы производится по некоторому заданному алгоритму и не требует много времени.
Обозначения.my_func - вычисляемая функцияМ - массив для аргументов функции my_funcF - массив для значений функции my_funcM_distr - распределенный массив для M (той же размерности)F_distr - распределенный массив для F (той же размерности)
Исходная программа
Результирующая программа
function test_1()
% инициализация данных
n=1000;
...
% резервирование памяти
M = zeros(10,n);
F = zeros(1,n);
% заполнение матрицы М
...
function test_1()
% инициализация данных
n = 1000;
...
% резервирование памяти
M = zeros(10,n);
F = zeros(1,n);
% заполнение матрицы М
...
% создание распределенных массивов
% разбиение по умолчанию проводится
% по столбцам (2-му измерению)
M_distr = codistributed(M);
F_distr = codistributed(F);
% вычисление функции
for i = 1:n
F(1,i) = my_func(M(:,i));
end
% вычисление функции
for i = drange(1:size(M_distr,2))
F_distr(1,i) = my_func(M_distr(:,i));
end
% сбор результатов на 1-ом процессе
F = gather(F_distr,1);
save ('test_res_1.mat', 'F', 'M');
if labindex == 1
save ('test_res_1.mat', 'F', 'M');
end
return
end
return
end
Пример 2. Построение массива из частей.
Распределенный массив как объединение локальных массивов
Сформировать таблицу значений функции 2-х вещественных переменных ((x,y) --> F) на прямоугольной сетке размера 120x200, заданной векторами (x1:x2:x3 и y1:y2:y3). Существует алгоритм вычисления значения функции в точке , обозначенный my_func.
Обозначения.my_func - вычисляемая функция 2-х переменныхF - массив для значений функции my_funcF_loc - локальный массив, который заполняется соответствующими значениями функции на каждом процессе и затем берется за основу (рассматривается как сегмент) при построении распределенного массива F_distrF_distr - распределенный массив, содержимое которого собирается в массиве F на 1-ом процессе
Вариант 1.
Распараллеливание проводится по внешнему циклу (по x - первому индексу).
Предполагается, что число строк 120 кратно numlabs - числу процессов, заказанных при запуске программы на счет.
Исходная программа
Результирующая программа
function test_2()
% инициализация данных,
% в частности,
% x1, x2, x3, y1, y2, y3
...
function test_2()
% инициализация данных,
% в частности,
% x1, x2, x3, y1, y2, y3
...
m_x = x1:x2:x3;
n = 120/numlabs;
% выделение памяти
F = zeros(120,200);
% вычисление функции
i = 1;
for x = x1:x2:x3
...
% выделение памяти
F_loc = zeros(n,200);
% вычисление функции
% i = 1;
for i = 1:n
k = (labindex-1)*n + i;
x = m_x(k);
...
j = 1;
for y = y1:y2:y3
F(i,j) = my_func(x,y);
j = j + 1;
end
i = i + 1;
end
j = 1;
for y = y1:y2:y3
F_loc(i,j) = my_func(x,y);
j = j + 1;
end
% i = i + 1;
end
% распараллеливание проведено по строкам
% (1-му измерению) => codistributor1d(1, ...)
codist = codistributor1d(1, [], [120 200]);
F_distr = codistributed.build(F_loc, codist);
F = gather(F_distr, 1);
save ('test_res_2.mat', 'F');
if labindex == 1
save ('test_res_2.mat', 'F');
end
return
end
return
end
Вариант 2.
Распараллеливание проводится по внутреннему циклу (по y - второму индексу).
Предполагается, что число столбцов 200 кратно numlabs - числу процессов, заказанных при запуске программы на счет.
Исходная программа
Результирующая программа
function test_2()
% инициализация данных,
% в частности,
% x1, x2, x3, y1, y2, y3
...
function test_2()
% инициализация данных,
% в частности,
% x1, x2, x3, y1, y2, y3
...
m_y = y1:y2:y3;
n = 200/numlabs;
% выделение памяти
F = zeros(120,200);
% вычисление функции
i = 1;
for x = x1:x2:x3
...
j = 1;
for y = y1:y2:y3
% выделение памяти
F_loc = zeros(120,n);
% вычисление функции
i = 1;
for x = x1:x2:x3
...
% j = 1;
for j = 1:n
k = (labindex-1)*n + j;
y = m_y(k);
F(i,j) = my_func(x,y);
j = j + 1;
end
i = i + 1;
end
F_loc(i,j) = my_func(x,y);
% j = j + 1;
end
i = i + 1;
end
% распараллеливание проведено по столбцам
% (2-му измерению) => codistributor1d(2, ...)
codist = codistributor1d(2, [], [120 200]);
F_distr = codistributed.build(F_loc, codist);
F = gather(F_distr, 1);
save ('test_res_2.mat', 'F');
if labindex == 1
save ('test_res_2.mat', 'F');
end
return
end
return
end
Замечание. Эту задачу, если памяти достаточно, можно запрограммировать и первым способом, т.е. путем деления большого массива на части. В этом случае распределение данных по процессам будет сделано автоматически (требование кратности исчезает).
Вверх
Как убедиться в работоспособности программы
Перед первым запуском программы-функции на кластере ее стоит проверить сначала в однопроцессорном варианте с отладчиком Debug (см.,например, Debug a MATLAB Program), а затем в параллельном режиме pmode. Поскольку политика использования вычислительных ресурсов ИММ УрО РАН не предполагает длительных вычислений на управляющем компьютере, запускать программу в режимах Debug и pmode следует с тестовым набором данных.
Итак, рекомендуется следующая последовательность выхода на счет:
Debug
pmode
кластер
Запуск параллельной программы с отладчиком Debug (в однопроцессорном режиме) можно осуществить, например, предварительно открыв текст программы в редакторе (Editor), установив необходимые контрольные точки (щелкая, к примеру, левой клавишей мыши справа от номера нужной строки) и нажав клавишу F5 (см. пункт меню Debug).
Стартовать режим pmode на 4-х процессах (для наглядности) можно в окне Matlab (Command Window) с помощью команды
pmode start 4Затем в командной строке открывшегося параллельного окна (Parallel Command Window) вызвать свою программу.
Закрыть параллельный режим можно или из Parallel Command Window командой
exitили из окна Matlab командой
pmode exitДля контроля за данными полезна функция getLocalPart.
Запуск на кластере можно осуществить в окне Matlab с помощью функции imm_sch.
Использование параллельного профилирования
Основные понятия
Профилирование предназначено для выявления наиболее затратных по времени мест в программе с целью увеличения ее быстродействия (см. Profile to Improve Performance).
Профилирование параллельных программ определяет как затраты на вычисление функций, так и затраты на коммуникации (см. Profiling Parallel Code).
Профилирование в Matlab осуществляется с помощью инструмента, называемого профилировщик или профайлер (profiler).
Результатом профилирования являются суммарный и детальный отчеты о выполнении программы.
Суммарный отчет о профилировании параллельной программы содержит для каждого параллельного процесса (lab) информацию о времени вычисления функций и частоте их использования, а также о времени, затраченном на обмены (коммуникации) и ожидание обменов с другими процессами.
Для проблемных функций полезно выдавать детальный отчет, в котором содержится статистика по строкам функций, а именно: время выполнения и частота использования.
Код программы можно считать достаточно оптимизированным, если в результате изменения алгоритма большая часть времени выполнения программы будет приходиться на обращения к нескольким встроенным функциям.
Вверх
Действия пользователя
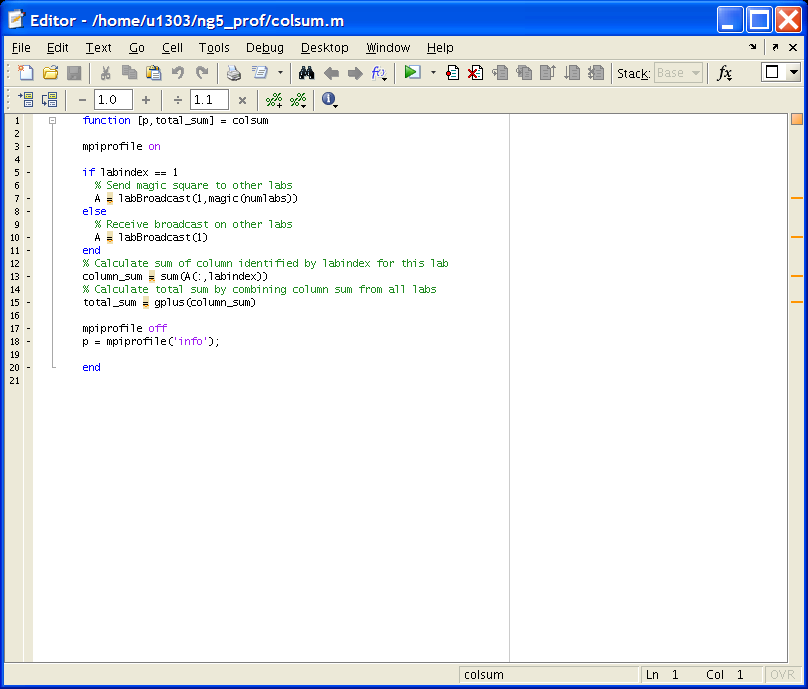
Действия пользователя иллюстрируются на примере функции colsum() по аналогии с функцией foo() из раздела Examples описания команды (функции) mpiprofile.
Внесение изменений в текст программы
1) В заголовок функции добавить выходной параметр для информации о профилировании, назовем его p:
function [p, total_sum] = colsum2) В тело функции добавить команду mpiprofile, а именно:
mpiprofile onдля включения профилирования (начала сбора данных)
mpiprofile offдля выключения профилирования при необходимости (или желании)
p = mpiprofile('info');для доступа к сгенерированным данным и сохранения собранной информации в выходном параметре p.
Пример скорректированной программы приведен ниже в файле ml_prof_colsum.PNG.
Запуск программы на профилирование
1) При запуске функции указать увеличенное соответственно на 1 число выходных параметров.
2) Запустить программу-функцию из командной строки:
mlrun -np 9 -maxtime 5 colsum '2'или в окне Matlab:
j = imm_sch(9,5,@colsum,2);Замечание. Окончания счета при желании можно ждать с помощью команды wait(j).
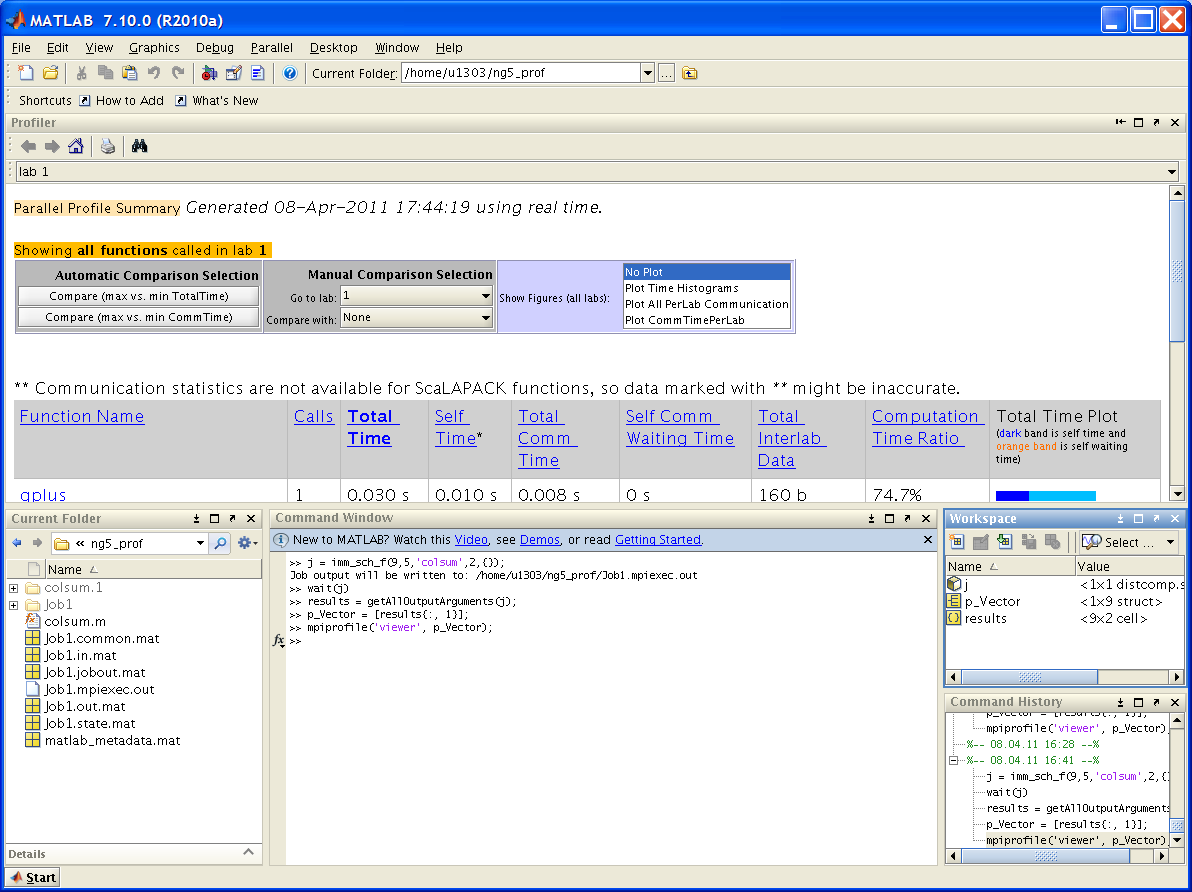
Просмотр результатов профилирования
Пусть программа запущена в окне Matlab. Тогда по окончании счета следует
1) получить результаты:
out = j.fetchOutputs;2) выделить вектор с информацией о профилировании:
p_vector = [out{:, 1}];3) запустить вьюер:
mpiprofile('viewer', p_vector);Подобные действия для ранних версий приведены ниже в файле ml_prof_session.PNG.
Для повторного использования полученные переменные можно сохранить командой:
save ('res_prof', 'out', 'p_vector')(в файле res_prof.mat) и загрузить при необходимости в другом сеансе:
load ('res_prof')
Вверх
Некоторые советы
Первая реализация программы должна быть настолько проста, насколько возможно, т.е. не рекомендуется ранняя оптимизация.
Профилирование может быть использовано
- в качестве инструмента отладки;
- для изучения (понимания) незнакомых файлов.
При отладке программы детальный отчет даст представление о том, какие строки выполнялись, а какие нет. Эта информация может помочь в расширении набора тестов.
Для очень длинного файла с незнакомым матлабовским кодом можно использовать профайлер, чтобы посмотреть, как файл в реальности работает, т.е. посмотреть вызываемые строки в детальном отчете.
При использовании профилирования копию 1-го детального отчета рекомендуется сохранить в качестве основы для сравнения с последующими, полученными в ходе улучшения программы.
Замечание. Существуют неизбежные отклонения времени, не зависящие от кода, т.е. при профилировании идентичного кода дважды можно получить слегка различные результаты.
| Прикрепленный файл | Размер |
|---|---|
| 38.44 КБ | |
| 85.25 КБ |
{kind=link}
{kind=link}
О реализации работы системы Matlab на кластере
Запуск параллельной программы пользователя на кластере организован в соответствии с рекомендациями разработчиков Matlab в разделах
Programming Distributed Jobs и
Programming Parallel Jobs
с учетом используемого в ИММ планировщика (типа Generic Scheduler в терминологии Matlab).
Замечание. Ссылки на сайт разработчика Matlab даны на момент подключения системы Matlab (~2010).
Для осуществления такого запуска необходимо:
Со стороны системы Matlab
1) Обеспечить наличие функций Submit и Decode.
Функция Submit выполняется Matlab клиентом на хосте (управляющей машине) и основное ее назначение - установить необходимые переменные окружения, в том числе для функции Decode, перед обращением к планировщику (Scheduler).
Функция Decode выполняется автоматически Matlab сервером (worker) на каждом узле и основное ее назначение – забрать переданные планировщиком через переменные окружения значения, которые Matlab worker использует для проведения вычислений на узлах кластера.
2) Задать последовательность действий для описания параллельной работы (Job, точнее ParallelJob), в рамках которой и будут проводиться программой Matlab вычисления функции пользователя на кластере. Основные действия описаны на сайте Matlab и продемонстрированы, в частности, на примере. В ИММ с учетом пакетной обработки эти действия дополнены перехватом stdout и stderr, т.е. установкой свойства CaptureCommandWindowOutput в значение true для всех задач (Task) программируемой работы (Job) (коротко о Job и Task см. в пункте Основные сведения).
С целью дальнейшего многократного использования все необходимые для программирования параллельной работы действия были оформлены в виде служебной функции imm_sch_f (позднее переименованной в imm_sch), обозначенной на рисунке ниже как scheme function. Этой функции в качестве параметров передаются число параллельных процессов, время счета, имя и параметры функции пользователя, предназначенной для распараллеливания вычислений.
Функция imm_sch_f служит для запуска параллельных программ.
При запуске параллельной программы в окне системы Matlab пользователь явно обращается к этой функции.
3) Для своевременного удаления информации о завершенных работах предоставить в распоряжение пользователя функцию job_destroy.
Со стороны планировщика
1) Обеспечить пользователя командой запуска параллельной программы из командной строки, аналогичной mpirun (заменённой впоследствии на mqrun). Эта команда получила название mlrun. При вызове mlrun пользователь указывает число необходимых для счета процессов, максимальное время счета и имя, а при наличии и параметры, программы-функции. Команда mlrun в интерактивном текстовом режиме (-nodisplay) вызывает программу matlab для выполнения функции imm_sch_f (scheme function на рисунке), передавая ей все необходимые аргументы.
2) Обеспечить наличие скрипта, реализующего механизм постановки в очередь программы пользователя, сформированной для запуска на кластере в рамках Matlab. Этот скрипт назван matlab_run.
Используем предлагаемую на сайте Matlab схему запуска программы пользователя на кластере для демонстрации получаемой у нас цепочки вызовов (для определенности на um64 в системе пакетной обработки заданий СУППЗ, 2009 г.).
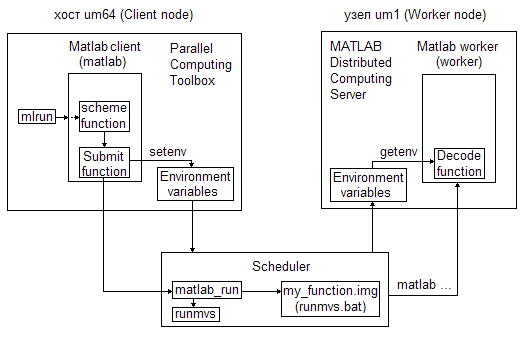
Источниками для написания собственных функций и скриптов послужили:
1) Ориентированные на Generic Scheduler примеры:
- функций Submit и Decode,
- схемы программирования параллельной работы на хосте в клиентской части Matlab и
- действий по удалению (разрушению, destroy) законченной и ненужной уже работы (Job).
2) Документация (скрипты типа pbsParallelWrapper) в каталогах вида:
/opt/matlab-R2009b/toolbox/distcomp/examples/integration/{pbs|ssh|lsf} где R2009b - название версии
3) Скрипты запуска, такие как mvarun_p2 из каталога
/common/runmvs/binСписок наших служебных функций и скриптов приведен в таблице:
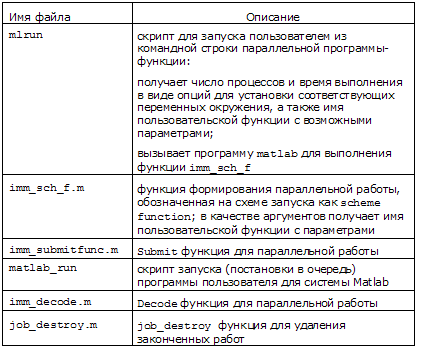
При переходе на новую версию Matlab, например R2010a, следует
- добавить одноименные ссылки в каталог /opt/matlab-R2010a/toolbox/local
на наши служебные Matlab функции, хранящиеся в каталоге /opt/matlab-imm, а именно: imm_sch_f.m, imm_submitfunc.m, imm_decode.m, job_destroy.m ;
- скорректировать файл /etc/profile.d/matlab.sh.